- Research
- Open access
- Published:
A European proposal for quality control and quality assurance of tandem mass spectral libraries
Environmental Sciences Europe volume 32, Article number: 43 (2020)
Abstract
Background
High resolution mass spectrometry (HRMS) is being used increasingly in the context of suspect and non-targeted screening for the identification of bioorganic molecules. There is correspondingly increasing awareness that higher confidence identification will require a systematic, group effort to increase the fraction of compounds with tandem mass spectra available in central, publicly available resources. While typical suspect screening efforts will only result in tentative annotations with a moderate level of confidence, library spectral matches will yield higher confidence or even full confirmation of the identity if the reference standards are available.
Results
This article first explores representative percent coverage of measured tandem mass spectra in selected major environmental suspect databases of interest in the context of human biomonitoring, demonstrating the current extensive gap between the number of potential substances of interest (up to hundreds of thousands) and measured spectra (0.57–3.6% of the total chemicals have spectral information available). Furthermore, certain datasets are benchmarked, based on previous efforts, to show the extent to which acquired experimental data were comparable between laboratories, even with HRMS instruments based on different technologies (i.e., quadrupole–quadrupole-time of flight versus ion trap/quadrupole-Orbitrap). Instruments and settings that are less comparable are also revealed, primarily linear ion trap instruments, which show distinctly lower comparability.
Conclusions
Based on these efforts, harmonization guidelines for the acquisition and processing of tandem mass spectrometry data are proposed to enable European (and ideally worldwide) laboratories to contribute to common resources, without requiring extensive changes to their current in house methods.
Background
Detection, annotation and identification
The goal of suspect and non-targeted analysis is to provide extensive qualitative information on the chemical composition of a sample. These analytical approaches are possible because of recent technologies and instrumentation, which are capable of generating large amounts of chemical information from low sample amounts. In particular, high-resolution mass spectrometry (HRMS) is one of these innovative technologies for large-scale and high-throughput profiling of complex samples [1]. However, assigning chemical identities to a set of mass spectrometric signals is not trivial and requires strongly consolidated data processing as well as appropriate quality assurance (QA) and quality control (QC) procedures. Particularly the confidence of the assigned chemical identities (annotations) is a crucial issue [2]. As the aim is typically to confirm the presence of as many compounds as possible, adequate strategies have to be used in reporting the results, both for research and in the context of regulatory use. While QA/QC aspects are well established in the field of conventional targeted methods, these are currently less developed for non-targeted analyses, although these are discussed actively in both the metabolomics and environmental communities (e.g., [2, 3]).
This article explores possible strategies for QA/QC of tandem mass spectral libraries and databases, especially suitable for the annotation of chemicals of emerging concern in the context of human biomonitoring (specifically within the Human Biomonitoring for Europe project, HBM4EU, http://www.hbm4eu.eu) and environmental monitoring (initiatives originating from the NORMAN Network, http://www.norman-network.net). It reflects on existing initiatives and approaches that can be used to assign well-defined confidence levels to annotated biomarkers (either of exposure or effect), to check the quality of existing sets of tandem mass spectra data, and for acquiring new experimental data, as well as any gaps that may need to be addressed.
For this article, a few definitions are clarified here, with further definitions given in the Glossary below. Detection refers to the collection of compound-specific data by instrumental analysis. For chromatography coupled to mass spectrometry, this collected data may include retention times, mass-to-charge ratios (m/z) of molecular ions, adducts and possibly fragment ions as well as the presence and relative abundances of fragment ions and isotopologues. Annotation is the act of linking a detected mass spectrometric feature with a chemical identity, taking into account the detected chromatographic and spectrometric characteristics. Identification is the process of proving or verifying that the annotated compound is indeed the proposed chemical (i.e., the annotation can be confirmed). Annotation is generally performed using analytical evidence of the measured dataset alone [1,2,3], along with additional supporting evidence (e.g., experimental context [2] and metadata [4,5,6]). In contrast, identification is generally accomplished by comparing measured data sets (e.g., using reference standards), where one set of features is obtained from the analysis of an unknown compound; the other from a reference standard of known identity. In this context, defining objective metrics for confirmation of identity is a challenging task. Ideal metrics should minimize the number of false positive and false negative identifications (see below).
Sufficient analytical data must be available to enable definitive identification. Like a fingerprint, this evidence should be a unique set of information capable of excluding all other chemical entities from consideration (unequivocal identification). In the case of a mass spectrometry (MS)-based characterization, such a chemical fingerprint can partly be created in silico (e.g., m/z-values of molecular ions, relative abundances of isotopologues given the molecular formula and, to some extent, fragment ions) and/or by analyzing reference standards (e.g., m/z-values of fragment ions, retention times in chromatographic systems). These “fingerprints” (often specific for distinct instrumental settings) are generally stored in databases. While predictive (in silico) methods exist for both fragment (e.g., [7, 8]) and retention time information (e.g., [9,10,11,12]), these are not yet sufficiently accurate for unequivocal identification, although these are constantly improving in accuracy. Thus, complete “standards-free” identification is not yet possible for HRMS, although it is becoming increasingly possible to get reasonable annotations using “standards-free” approaches.
From an analytical point of view and depending on the available information for annotation, chemicals can be divided into three categories, which we define as follows for this article (see Fig. 1).

The chemical space difference between targets, suspects and non-targets/unknowns
Targets are compounds (“knowns”) that are preselected for analysis in a sample and for which full mass spectrometric reference data, including MS/MS fragmentation and retention time, is available for annotation. The reference data are usually acquired with certified reference standards in house; the reference mass spectra and, in some cases retention times (depending on the database), are stored in mass spectral databases.
Suspects are known compounds (“known unknowns” [13]) that are expected (“suspected”) to be present in a sample, but for which either no reference standard (in house) or incomplete mass spectrometric reference data are available, such that unequivocal annotation is not always directly possible. In suspect screening, this could be either missing or measured with alternative methods, or predicted with computational tools, and as such not of sufficient accuracy in many cases to allow reliable annotation—although it may provide supporting evidence. For instance, while MS information can be computed reliably from the structure, the in silico prediction of MS/MS fragmentation and retention time information still needs to be improved.
Both targets and suspects represent subsets of the entire chemical space in a sample (Fig. 1). Suspects can be “converted” into targets by collecting comprehensive mass spectrometric reference data that enables unequivocal identification of the suspect compound (usually reliant on the availability of reference standard compounds). The remaining signals in the sample are generally termed non-targets or unknowns—for which no target or suspect identity can be assigned readily. These require full elucidation and are beyond the scope of this article.
Inspired by European regulatory documents and a classification system originally proposed by Sumner et al. [14] for metabolomics, a classification system tailored for HRMS primarily for the environmental context was proposed by Schymanski et al. [2] in 2014. Level 1 (confirmed structure) described identification that has been verified via the appropriate measurement of a reference standard with MS, MS/MS and retention time matching, matching the definition of targets above. A “probable structure” (Level 2) is obtained by unambiguous matching literature or library data (Level 2a) or via diagnostic evidence (Level 2b), where the diagnostic evidence must clearly rule out all other candidate structures. “Tentative candidates” (Level 3) describe the case where the available data provide evidence for possible or likely structure(s), but insufficient information exists for one exact structure only (e.g., positional isomers). Level 4 or 5 identifications are typically “unknowns”, where only the molecular formula (Level 4) or exact mass (Level 5) are known. Since initiation of the level system in 2014, several practical cases have evolved within each level and these are, for instance, now encoded into the mass spectral processing software RMassBank [15].
Tandem mass spectral databases: current status
Tandem mass spectral databases are indispensable tools for compound annotation in non-targeted HRMS workflows based on soft-ionization mass spectrometry (typically liquid chromatography (LC)-HRMS) and good matches can yield Level 2a annotations in many cases. Several reviews are available describing the development and application of tandem mass spectral databases [3, 16,17,18,19,20,21,22]. Typically, a tandem mass spectral database represents an organized collection of tandem mass spectral data within a management system. The database management system enables the user, or other applications, to interact with data within the database itself. Tandem mass spectral databases are acquired by the analysis of reference standards. Since a fragmentation spectrum can look different depending on the excitation process (e.g., resonant vs. non-resonant) as well as the collision energy applied to the parent ion, state-of-the-art databases include sets of compound-specific spectra that were acquired by applying different collisions energy settings, as well as different instruments [23, 24]. Fragmentation is typically accomplished by collision-induced dissociation (CID) or higher-energy collisional dissociation (HCD). Usually, the spectral information is processed prior to storage in a library. Curation efforts may include manual inspection of mass spectra by experienced mass spectrometrists, noise and artifact removal, recalibration of spectra and peak annotations, as well as inter-library comparisons [15, 23, 25,26,27]. In some databases, such as the Human Metabolome Database (HMDB [28]) and MassBank of North America (MoNA, [29]), experimental data are now complemented with in silico-generated spectra.
In 2016, the overlap of compounds with tandem mass spectra from authentic reference standards in most public and commercial databases was evaluated by Vinaixa et al. [16]. A total of 27,622 unique compounds were present across all databases. Among the 7127 compounds in the four open databases HMDB 3.0 [30], MassBank [24], the Global Natural Product Social Molecular Networking library (GNPS) [31], and the RIKEN MSn spectral database for phytochemicals (ReSpect) [32], only 18 compounds (< 1%) had at that time at least one form of spectral data in all databases. When comparing all combined open databases versus four commercial ones, only 225 compounds out of 27,622 (< 1%) had at least one form of spectral data in all databases. The ratio of compounds in each database with any type of spectral data in two or more databases was generally > 50%, with the exception of METLIN and GNPS, which only overlapped approximately 35% with other databases in terms of compounds. As there is a relatively low overlap of compounds among existing spectral databases, most scientists currently use multiple databases. Since the 2016 review, many of the databases have expanded their compound coverage immensely and many of the open libraries have cross-imported their spectral records. However, the issue of overlap and coverage of relevant substances in chemical space remains [17].
Conceptually, the premise of spectral library searching is very simple: the fragmentation pattern of a molecule is a reproducible fingerprint of that molecule under a given set of fixed conditions, such that unknown spectra acquired under similar conditions can be identified via spectral matching [33]. Automated spectral library searching involves software with tailor-made search algorithms for tandem mass spectral databases [34,35,36,37]. The search score obtained following the database search represents the likelihood that the searched spectrum corresponds to a given reference spectrum in the mass spectral database. A low score indicates that the experimental fragmentation pattern has low similarity to any stored reference spectrum. A high score indicates significant spectral overlap and, consequently, that the analyte is likely either structurally similar or even identical to the reference compound. Library search should be both sensitive and specific, producing as few false negative and false positive results as possible. Ideally, the scores obtained should be able to distinguish true and false positive matches [18]. To compare with historical targeted methods applied in a regulatory context (e.g., forensic toxicology and food safety), the primary objective of a screening method is to limit the risk of false negatives (e.g., 1%) and to keep to an acceptable risk of false positives (e.g., 5%). The latter should be further reduced by confirmatory analyses using reference standard compounds. Non-targeted methods with consolidated QA/QC thus have to consider and document these issues during the development and evaluation of method performances, as well as in reporting of results. False discovery rates, applied successfully in proteomics for many years [38], are now being developed for small molecule MS/MS [39], but are not yet widely integrated into tandem library software.
There is also extensive discussion about the robustness and transferability of tandem mass spectral libraries. For a long time, the predominant opinion was that libraries would only be useful on the instrument used to acquire reference spectra, due to the limited reproducibility of tandem mass spectra. This situation has changed thanks to both progress in instrument technologies and informatics tools. Databases combining advanced library designs with tailor-made search algorithms have been shown to enable reliable compound identification with spectra acquired in different laboratories with various instruments and different instrument settings [18, 27]. While pre-acquisition harmonization of analytical procedures was researched, the participating laboratories encountered a number of difficulties [40]. Thus, the current trend is rather to look for a post-acquisition flexibility of the MS/MS reference library and associated matching algorithms to deal (as much as possible) with the diversity of imported experimental data without sacrificing the ambitioned confidence level in terms of correct annotation.
Over the last 10 years, there has been substantial progress in the quality of tandem mass spectral databases. Today, spectral acquisition of reference spectra is accomplished regularly on high-resolution instrumentation (i.e., quadrupole–quadrupole-time of flight (QqTOF), Orbitrap) employing multiple collision energies for fragmentation to comprehensively cover the breakdown curves of reference compounds. Besides the protonated and deprotonated molecular ions, adduct ions, in-source fragments as well as isotopologues are commonly selected as precursor ions [18, 25, 26]. Furthermore, to improve spectral quality, generally only curated spectra are stored in databases, which come bundled with improved search algorithms. As knowledge and understanding of mass spectra increase, automated curation procedures are being implemented and constantly improved to reduce the manual curation load associated with mass spectral database creation [15, 25]. Overall, the ambition for a “universal tandem mass spectral database” is closer to a reality. However, this ambition requires definition and implementation of some common procedures to ensure the reliability and robustness of the generated data, both stored in the desired tandem mass spectral reference library and generated from each experimental sample.
A number of reference tandem mass spectral databases exist that have to be considered in the frame of identification of chemicals of emerging concern, in terms of structuration and/or content. These existing resources should serve as a basis to avoid unnecessary time spent in re-implementing existing and reliable elements, and to ensure a coherence of the ambitioned outputs with potential established standards. However, an obvious lack of high-level QA/QC consolidation appears within many of these existing databases (e.g., percentage of erroneous information, insufficiently or non-adequately curated spectra), together with some necessary adjustments for specific applications (e.g., human metabolites of contaminants are not well represented as compared to parent compounds).
Methods—proposed QA/QC framework
Generic strategy for converting suspects into targets
The long-term goal in terms of developing screening capabilities would be to progressively include a large part, ideally all, of the compounds listed in “suspect lists of interest” (e.g., [41]) into corresponding entries in tandem mass spectral libraries. This would allow the conversion of suspects (generally Level 3) into targets (Level 1, if the retention time information has been measured in house or on an identical chromatographic regime elsewhere) or higher confidence tentative matches (Level 2a, spectral library match). The strategy for accomplishing this aim involves (1) QC of already acquired tandem mass spectral data to determine how many suitably comparable mass spectral records exist and (2) QA-guided acquisition of new reference spectra, shown in Fig. 2.

The proposed workflow to convert suspects into targets
To determine a baseline for the status of environmentally and toxicologically relevant compounds and their presence in various resources, a mapping exercise was performed. Compound numbers (number of entries) were obtained for the CompTox Chemicals Dashboard [42], NORMAN SusDat [43], HMDB [28], DrugBank [44], the Toxic Exposome Database (TEDB) [45] and Exposome Explorer [46] from their respective websites or download files on March 15, 2019. CompTox numbers mapping to mzCloud [47], MassBank [48] and WRTMD [49] were obtained via downloading the respective list files (list codes on https://comptox.epa.gov/dashboard/chemical_lists are: MZCLOUD, HDXNOEX, MASSBANKEUSP, MASSBANKREF, MYCOTOXINS, WRTMD), also on March 15, 2019 and counting/merging by InChIKey first block (thus ignoring stereochemistry). HMDB MS/MS numbers were obtained from download files (March 15, 2019) and cross-checked with InChIKey mappings still on record from a previous study [16]; also counting by InChIKey first block. SusDat mappings to MassBank were obtained by extracting list S1 results from the download file. To provide a global, up-to-date overview, all compounds with MS/MS annotation that were listed in the PubChem [50] Table of Contents Browser (https://pubchem.ncbi.nlm.nih.gov/classification/#hid=72) were exported as SMILES [51], converted to InChIKeys using Open Babel [52], and counted by unique InChIKey first block. While both DrugBank and T3DB contain MS/MS records, this information is not available in their export files, and these contain high overlap with HMDB where the information is mapped extensively in the download files.
The authors note that while many more resources are available, these are open resources with pre-mapped information to the highest quality and relevant MS/MS records to form a sufficient information basis for the outcome of this article.
Quality control and benchmarking of tandem mass spectral libraries
The library of the Helmholtz Centre for Environmental Research (UFZ) being part of MassBank was used as test set to demonstrate the usefulness of the proposed strategy. The UFZ library (at that stage) contained 636 MS/MS spectra corresponding to 167 compounds. Reference spectra were recorded on a LTQ-Orbitrap XL (Thermo Fisher Scientific, Waltham, MA, USA). HCD product-ion spectra were acquired at three different collision energy levels (HCD 35, 55, 80) at a nominal resolving power of 30,000. The R package RMassBank was used to perform recalibration and clean-up of acquired spectra [15]. The curated spectra are available at https://github.com/MassBank/MassBank-data/tree/master/UFZ.
The spectra of the UFZ library were searched against “The Wiley Registry of Tandem Mass Spectral Data” (WRTMD) [53]. Library search was accomplished using ‘MSforID Search’ [34, 35] as described in the Additional file 1. The spectra of compounds covered in both UFZ and WRTMD served as positive controls. The number of positive identifications obtained with the positive controls were counted and used to calculate the statistical parameter sensitivity (= true positive rate).
Interlaboratory study to validate acquired reference spectra
An interlaboratory study was organized to verify that participating laboratories were generating new reference data with experimental settings and workflows that were compatible with existing reference spectra collections. Each participating lab was asked to use fifteen reference compounds for producing tandem mass spectral libraries. Table 1 includes the set of compounds used in this study. They have already been applied to test the transferability of the WRTMD in different laboratories with various available instruments and procedures (e.g., [18, 34, 36]). Seven laboratories involved in the NORMAN Network and/or HBM4EU participated in the interlaboratory study. An overview of the applied instrumentation as well as the applied fragmentation technique is provided in Table 2 as well as in the Additional file 1.
The seven collections of centroided, averaged and curated tandem mass spectra were benchmarked against the WRTMD. Benchmarking included two sets of experiments: (1) matching the test spectra to the WRTMD, and (2) matching the spectra of the 15 test compounds included in the WRTMD to modified libraries derived from the WRTMD by substituting the original reference spectra with the newly generated libraries. For statistical evaluation of library search performance, all test sets were grouped according to the collision energy settings used to acquire the individual spectra.
Results and discussion
Starting point—overview on existing tandem mass spectral data for chemicals of potential concern
Various initiatives and/or sources of information have documented proposed lists of chemicals of emerging concern in various contexts, mainly in the field of environment and toxicology. Examples are the many lists on the NORMAN Suspect List Exchange [41] and the corresponding merged database SusDat [43], the CompTox Chemicals Dashboard [42] and a series of topical databases from the Wishart laboratory and collaborators (e.g., HMDB [28, 30], DrugBank [44], the Toxin and Toxin Target Database (T3DB) or Toxic Exposome Database (TEDB) [45] and the Exposome Explorer database [46]). While these sources overlap to some extent, they also provide a lot of complementary information and functions. As several reviews have covered the number of overlapping substances in tandem mass spectral libraries in various contexts (especially metabolomics) recently [16, 17, 54], the focus here will be on substances of interest for the environmental and biomonitoring contexts, using the resources mentioned. Looking into these collections, tandem mass spectral fragmentation information is already available for a considerable number of suspects in MassBank, HMDB, WRTMD or mzCloud, but this represents only a fraction of the substances actually present in the respective resources.
An overview of the number of compounds in the respective resources, as well as the number of entries that map to mass spectral data within that resource is given in Fig. 3. The CompTox Dashboard (875,000 compounds) includes 3997 compounds in mzCloud, 2377 in MassBank and 1429 in WRTMD, corresponding with 5019 unique compounds (ignoring stereochemistry differences), thus 0.57% of the resource. HMDB (144,098 compounds) contains MS/MS data corresponding to 750 unique compounds (ignoring stereochemistry), or 0.66% of the resource. NORMAN SusDat contains 40,180 entries, of which 1387 are in MassBank (3.6% of SusDat). This overview shows that tandem mass spectral data is available only for a rather low number of compounds. A further complicating factor is that these tandem mass spectral data are spread among several spectral collections. For the vast majority of interesting suspects, no public mass spectral data exists and measured mass spectral data will have to be newly generated, if possible. While METLIN now claims MS/MS spectra of over 500,000 chemicals (https://metlin.scripps.edu/, accessed 8 Dec. 2019), information on the coverage is not available, nor are the spectra openly available. However, as the PubChem database [50] aggregates information from a number of sources, the 74,678 compounds with MS/MS annotations (ignoring stereochemistry; 89,726 with stereochemistry), of 102,404,298 compounds (~ 0.073% of PubChem) give a reasonable indication of the total number of compounds with MS/MS information available to some extent, although some of these are in silico and many of these are not directly relevant for human biomonitoring or environmental studies.

Selected resources relevant for environmental and/or human biomonitoring studies (in blue, see main text) as well as the corresponding mass spectral entries, where available (orange). Both SusDat and HMDB have mass spectral resources integrated (SusDat partially), while CompTox has lists mapped to spectral libraries, indicating availability and PubChem has a Table of Contents with the number of compounds containing MS/MS annotations. CompTox, HMDB, and SusDat have more extensive in silico entries available that have not been represented here. While both TEDB and DrugBank list MS/MS entries (measured and in silico), this information is not as easily accessible as HMDB and many entries overlap with HMDB
The generation of new reference spectra is considered to represent an important element for the successful and long-term establishment of non-targeted LC–MS. As a single laboratory will not have the necessary resources available to handle the huge number of suspects ahead, this challenge must be addressed as a group. For successful realization of multi-partner generation of reference spectra, harmonization of acquisition and processing strategies is essential. Related QA actions imply application of generally agreed best practice procedures and participation in interlaboratory studies (see below). However, even by joining forces with respect to manpower and instrumentation, there will be further challenges ahead, and these are related to prioritization of suspect lists and availability of the corresponding reference standards.
Existing techniques for prioritizing chemicals are generally based on risk assessment [55, 56], which involves assessment of exposure and hazard. Other useful criteria might represent detectability by analytical techniques (e.g., LC–MS with ESI in positive or negative ion mode), legal status, importance for a defined research project [1], or simply the availability of reference information [4, 5, 57].
As things are now, over the next years a steady increase of the number of chemicals of emerging concern included in tandem mass spectral libraries is expected. Already available spectral collections are considered to represent nuclei for even larger collections. Therefore, much effort should also be put into the QC of already acquired tandem mass spectral data to determine how many suitably comparable mass spectral records already exist (see below).
Quality control of MassBank collections
MassBank is an important collection of reference tandem mass spectra [24]. Currently, 45 collections are available on MassBank (https://massbank.eu/MassBank/RecordIndex) with more than 55,075 tandem mass spectra (of 76,037 spectra total) representing 14,297 compounds (15,988 stereoisomers) total (over all spectral types). In terms of compound coverage, there is significant overlap between MassBank and the WRTMD that can be used to create sets of positive controls for testing the libraries.
Positive controls are particularly suitable for testing the quality and comparability of databases. Matching positive controls is used to determine the sensitivity (= true positive rates) of a database. Ideally, the obtained sensitivity values should be close to 100%. Negative controls are used to test the specificity (= true negative rate) of a database.
Initial benchmarking efforts between the Swiss Federal Institute of Aquatic Science and Technology (Eawag) MassBank collection and the WRTMD were published recently [27]. Spectra from the 233 overlapping substances between the two collections were used as positive controls. Of particular interest was the fact that the Eawag spectra were acquired with an Orbitrap instrument (HCD and CID), whereas the WRTMD spectra were acquired on a QqTOF. Spectra in the range of collision energy 20–50 eV on the QqTOF and 30–60% NCE on the Orbitrap provided optimal library matching results with sensitivity-values 95.1–98.4% [27]. Therefore, it was concluded that both collections enable reliable compound identifications, and that they are ready for use in suspect screening applications.
Another important spectral collection within MassBank is the UFZ library. The library contains tandem mass spectra of 167 compounds. The spectra were acquired on an Orbitrap with HCD. For each compound, reference spectra were acquired with three different collision energy settings. All spectra were curated and recalibrated before storing in MassBank. 87 reference compounds included in the UFZ library were also covered by the WRTMD. For each of these compounds two to eight spectra acquired at different collision energy settings (35, 55, 80%) were available. The corresponding 352 spectra represented positive controls suitable for QC of the UFZ library. The spectra were matched to the WRTMD and the number of positive matches was statistically evaluated (Fig. 4). The overall sensitivity was 89.7%. For 70 compounds, all test spectra performed well (amp > 5.0) and led to a positive match. There were, however, 16 compounds, of which at least one test spectra retrieved an amp-value below the specified threshold of 5.0 indicating insufficient similarity between test and reference spectra. Communicating the benchmarking results to the authors of the library initiated a fruitful discussion that also included reviewing of the raw data. This process identified reasons for the observed differences between test and reference spectra. In this way, the issues could be resolved and the corresponding entries in MassBank were updated.

Overview on the average match probability (amp) distribution obtained from matching 352 positive controls representing 87 compounds part of the UFZ library to the WRTMD. An amp-threshold of 5.0 was used to distinguish between positive and negative matches. The overall sensitivity was 89.7%. For 70 compounds, all test spectra led to positive matches with amp>5.0. For sixteen compounds at least one test spectrum retrieved an amp-value below the specified threshold of 5.0 indicating insufficient similarity between test and reference spectra
Thorough quality control of already existing spectral collections is able to identify libraries (or subsets thereof) for immediate application to compound identification in suspect screening. Likewise, benchmarking of tandem mass spectral libraries is a suitable approach to identify specific errors like low signal-to-noise ratios, improper mass calibration or wrong compound labeling. In this way, low-quality spectra can be identified, corrected or even deprecated.
Recommendations for QC of existing tandem mass spectral libraries
On the basis of the above results, as well as the previously published and discussed benchmarking studies, the following two-step QC procedure could be drafted and adopted:
Firstly, tandem mass spectral data should meet the following quality criteria:
- QC1:
Acquisition: High-resolution instrumentation (e.g., QqTOF, Orbitrap, Fourier Transform ion cyclotron resonance (FTICR)), typically with a minimum resolution of 10,000 in MS/MS mode, and m/z error lower than 10 ppm, to ensure contributions of broadly applicable spectra can be made from many laboratories.
- QC2:
Ionization: Positive and/or negative mode with a specified ionization technique (ESI, atmospheric pressure chemical ionization or atmospheric pressure photo ionization), as these are the most common methods.
- QC3:
Precursor ion isolation: The isolation window should be as narrow as possible to avoid fragmentation of multiple precursors, including isotopic peaks.
- QC4:
Fragmentation: Either Orbitrap-HCD or QqTOF-CID should be used for generating MS/MS spectra, as such spectra can be searched with higher sensitivity (see discussion below and Fig. 5).
Fig. 5 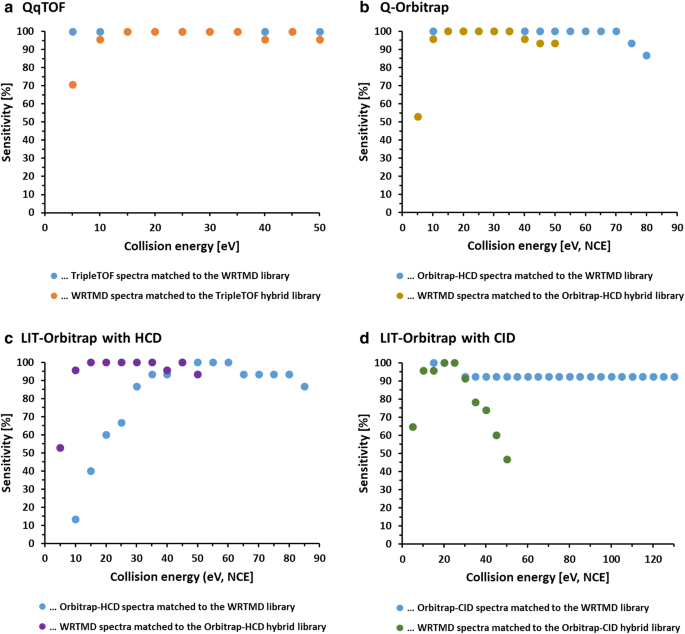
The reliability of library matching with the WRTMD and libraries derived from the WRTMD by substituting the original reference spectra with instrument-specific library spectra is shown by sensitivity versus collision energy for a a QqTOF instrument, b a Q-Orbitrap instrument with HCD, c a LIT-Orbitrap instrument with HCD, and d a LIT-Orbitrap instrument with CID, respectively. The collision energies are given in eV for the QqTOFs and NCE for the Orbitraps. a–c Reliable matches in a wide CE range, while d shows that the optimal CE window is smaller for LIT-Orbitrap instrument with CID
- QC5:
Mass range: Ideally the mass range should start at m/z ≤ 50 (instrumental limitations may preclude this, e.g., for instruments relying on ion trapping) wherever possible to include also small mass fragments. The acquired mass range should be given to avoid poor spectral matches due to the presence/absence of low m/z fragments in fragmentation spectra acquired with different scan ranges (see discussion below and Fig. 6).
Fig. 6 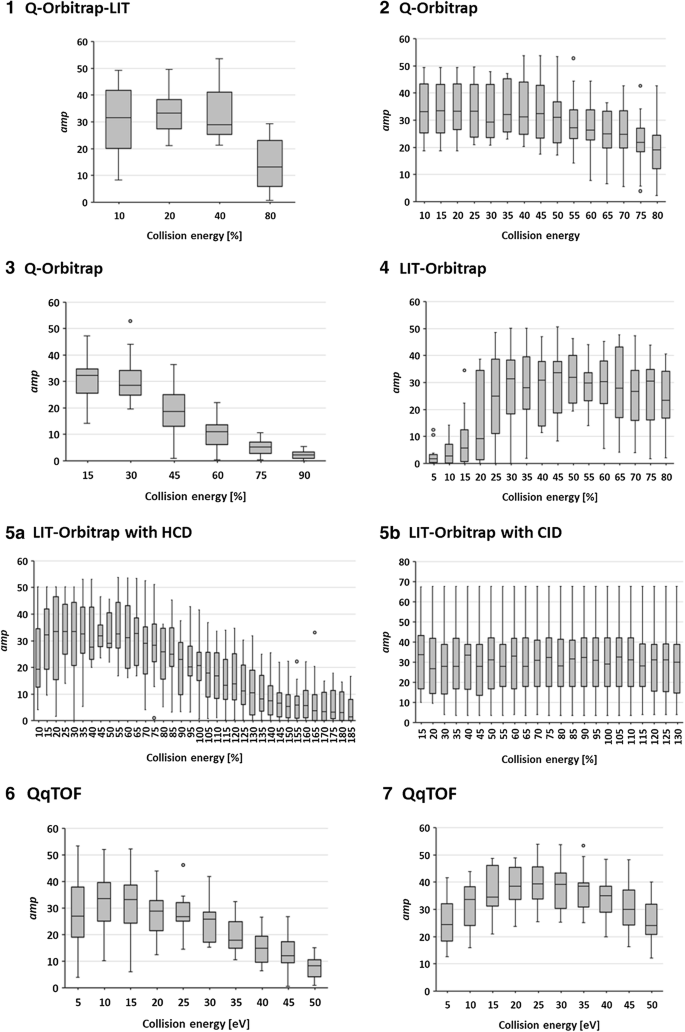
Overview on the average match probability (amp) distribution obtained from library matching of the spectral collections acquired during the interlaboratory study on the following instrument configurations: (1) Q-Orbitrap-LIT, (2, 3) Q-Orbitrap, (4) LIT-Orbitrap, (5a) LIT-Orbitrap with HCD, (5b) LIT-Orbitrap with CID, and (6, 7) QqTOF. The numbering (1–7) decodes the laboraties. Fragmentation on the Orbitraps 1–4 was accomplished with HCD. The collision energies are given in eV for the QqTOFs and NCE for the Orbitraps. The WRTMD served as reference library. Even for instruments with identical configurations a considerable inter-instrument variability in the optimal collsion energy range necessary for obtaining library matches with high amp-values was observed
- QC6:
Collision energies: Multiple collision energies (minimum 3) should have been recorded wherever possible over a meaningful range (e.g., 5 to 60 eV CID; NCE 10–60% HCD [27]) to form compound-specific breakdown curves.
- QC7:
Curation: Centroiding, filtering, noise removal, and recalibration should have been performed where possible to provide the best quality reference spectra.
- QC8:
Expert review: This is necessary to identify issues, such as artifacts, improper noise removal, or truncated spectra, which cannot always be captured automatically.


Secondly, spectral collections satisfying these conditions will proceed to the benchmarking step, using procedures described in the section “Quality Control of Mass Bank Collections”.
Multi-partner acquisition of new tandem mass spectral data
Interlaboratory harmonization studies are useful to verify that a laboratory is generating new reference data with experimental settings and workflows that are compatible with existing reference spectra collections. One way to characterize this interlaboratory comparability is to introduce a number of predefined known compounds and accompanying QA/QC criteria, such that these compounds must be detected and successfully identified with a given instrumentation and related procedure to validate the method appropriateness and reliability.
Seven laboratories involved in the NORMAN Network and/or HBM4EU project participated in the first harmonization study. The study was aimed to demonstrate compatibility and transferability of newly acquired tandem mass spectral data among participating laboratories as well as with already available reference spectra collections. The study involved the measurement of 15 reference standards (Table 1) on three different Orbitrap configurations (at four locations) and two QqTOFs (Table 2). The WRTMD served as the reference library.
The eight collections of centroided, averaged and curated tandem mass spectra were benchmarked against the WRTMD (Table 3). In a first set of experiments, acquired tandem mass spectra were matched to the WRTMD. The number of positive identifications obtained with individual test sets ranged from 73.2 to 100%. To prove that even the sets that showed a significant number of negative matches (e.g., laboratories 3, 4, and 5) contained suitable collision energy windows, the eight test sets were further grouped according to the collision energy settings used to acquire the individual spectra. As expected, a considerable number of subgroups were identified that led to 100% correct positive identifications (Fig. 5). The collision energy windows that appeared to be suitable for acquiring test spectra spanned at least 15 units (eV or % NCE). In the second set of benchmarking experiments, the spectra of the 15 test compounds included in the WRTMD were matched to eight libraries derived from the WRTMD by substituting the original reference spectra with the newly generated sets of reference spectra. The number of positive identifications obtained with individual test sets ranged from 78.5 to 99.5%. Also in this case, the test sets were further grouped according to the collision energy settings used to acquire the individual spectra. Like in the other experiment, a considerable number of subgroups were identified that led to 100% correct positive identifications (Fig. 5). The collision energy windows that appears to be suitable for acquiring library spectra spanned at least 10 units (eV or % NCE).
The interlaboratory study clearly demonstrated that the participating laboratories are able to acquire high-quality reference spectra for building libraries, while also providing further evidence that Orbitrap-HCD and QqTOF-CID introduce quite similar fragmentation reactions (Fig. 5). Thus, libraries produced on these types of instruments will offer complementary identification possibilities. Of utmost importance was the observation that there is a significant overlap of the compound-specific collision energy ranges between instruments (Fig. 5). Thus, databases that contain series of multiple spectra acquired on one instrument will enable reliable compound identifications when querying spectra from other instruments. Clearly, databases produced in different laboratories will offer complementary identification possibilities.
Another important result of the interlab study was the observation that even for instruments with identical configurations, a considerable inter-instrument variability in the optimal collsion energy range necessary for obtaining library matches with high match probability was observed (Fig. 6). Taking into consideration that all Orbitrap technology-based instruments were provided by the same manufacturer, a higher degree of similarity between those instruments regarding compound-specific breakdown curves was expected. The observation suggests that even after years of instrument development and optimization, harmonization of collision energy values has hardly been accomplished yet. The good news is, however, that state-of-the-art tandem mass spectral databases can cope with spectral variability leading to high reliability of a library match. The analyst applying these libraries just needs to find the optimal collision energy corridor for acquiring test spectra. The herein presented interlaboratory study could represent an appropriate strategy for this purpose.
The interlaboratory study also highlighted some limitations. These are mainly connected with the use of tandem-in-time fragmentation in the ion trap of the linear ion trap (LIT)-Orbitrap instrument (Fig. 5d). In contrast to quadrupole collision or HCD cells that use non-resonant CID, ion traps use resonant CID (i.e., several low-energy collisions during a longer time than non-resonant), which enables to produce fragmentation trees beyond MS2. Generally, these fragmentation trees cover the full range of possible fragmentation pathways, and are therefore specific identifiers for the corresponding molecules, which can be stored in databases (e.g., mzCloud [47]). With ion trap MS2, only parts of the entire range of possible fragmentation reactions are covered, even when applying higher collision energies [58, 59]. Such spectra match the lower energy part of spectral series acquired on tandem-in-space instruments (Orbitrap-HCD and QqTOF-CID) well. There is, however, limited overlap with spectra acquired at higher collision energies. Another problem of ion trap fragmentation is related to the “low mass cut-off”, or the so-called “1/3 rule” [59]. This means that fragment ions with an m/z-value below 1/3 of the m/z-value of the precursor ion are not trapped under normal operation conditions and are lost. Thus, a considerable part of fragment ions that are observed with higher collision energy of fragmentation on tandem-in-space instruments is not detectable with IT analysers. Thus, in comparison to Orbitrap-HCD and QqTOF-CID, Orbitrap-CID spectra are truncated. This truncation can hamper compound identification if abundant fragment ions are missing. One such example is desipramine (Fig. 7). At low collision energies, this compound has one abundant fragment ion at m/z 72.0808. This ion was not observed in the LIT-Orbitrap spectra since it displayed a m/z-ratio lower than 1/3 of the precursor ion (m/z 267 for [M + H]+). Accordingly, spectral match gave a low score.

The influence of the “1/3 rule” for ion trap spectra, exemplified with desipramine using a spectrum acquired on a LIT-Orbitrap instrument with CID and a QqTOF spectrum taken from the WRTMD. The black dot indicates the precursor mass isolated for MS/MS fragmentation (hollow dot). Due to the “low mass cut-off” observed on LIT-Orbitrap instruments, an abundant fragment ion is missing in the corresponding fragment ion mass spectrum
Another limitation of tandem mass spectral databases is highlighted in Fig. 8. It is well recognized that stereoisomers can hardly be distinguished from each other by tandem mass spectral fragmentation [13, 60]. But even the differentiation of constitutional isomers can be challenging. Fragmentation of such compounds may lead to identical products. In the worst-case scenario, tandem mass spectra will be identical. One such example of a pair of constitutional isomers comprises phentermine and methamphetamine. At higher collision energy levels, the fragmentation mass spectra of these two compounds show two identically intense fragment ions (Fig. 8), leading to ambiguous library search results.

An example of near identical tandem mass spectra: phentermine- and methamphetamine spectra acquired on a QqTOF instrument. The spectra were taken from the WRTMD. Black dots indicate precursor masses that triggered the MS/MS spectra (hollow dot). At higher collision energy levels, the fragmentation mass spectra of these constitutional isomers show two identically-intense fragment ions, leading to ambiguous library search results. Cases such as these demonstrate that library matching has to be complemented by orthogonal information such as retention time for higher identification confidence
QA recommendations for acquisition and processing of tandem mass spectral reference data
The results of the interlaboratory study, as well as the available experience and knowhow in building tandem mass spectral libraries, formed the basis for drafting and adopting the following recommendations:
- QA1:
Acquisition: High-resolution instrumentation (e.g., QqTOF, Orbitrap, FTICR) should be used for the acquisition of reference tandem mass spectra.
- QA2:
Instrument performance: Instruments should be properly tuned and calibrated (ideally daily or before commencing a batch analysis). High mass accuracy should be maintained using a lock mass or similar. The instrument should be capable of a minimum resolution of 10,000 in MS/MS mode, and the m/z error should be lower than 10 ppm.
- QA3:
Standards: Certified reference standards should be used to ensure that spectra will represent the linked structure.
- QA4:
Sample introduction: Samples may be introduced by direct infusion, flow injection or chromatography. A special caution should be paid to the minimal number of acquisition points (related to dwell time values and scan speed capabilities) to ensure a sufficient number of spectra for averaging. The possible occurence of background interferences should be checked by introducing blank samples.
- QA5:
Separation of mixtures: If reference compounds are introduced in mixtures, proper separation of the individual precursor ions (either during sample introduction or the mass spectrometric analysis) must be ensured to avoid the acquisition of chimeric spectra.
- QA6:
Dealing with isobars: If reference spectra are acquired in batches, isobaric compounds must not be processed consecutively, to avoid interferences due to carryover effects resulting in chimeric spectra.
- QA7:
Alternative precursors: While the primary precursors of interest may be protonated or deprotonated molecules, for some molecules other abundant signals corresponding to in-source fragments, isotopic peaks or other related species might be considered as additional precursor ions.
- QA8:
Isolation width: The precursor isolation window should be as narrow as possible to avoid fragmentation of multiple precursors, including isotopic peaks.
- QA9:
Fragmentation: Fragmentation should be accomplished by tandem-in-space techniques (e.g., HCD for Orbitrap, CID for QqTOF). As shown in Fig. 7, CID fragmentation with tandem-in-time instruments may produce spectra with limited overlap to spectra acquired with tandem-in-space techniques at higher collision energies. Another problem of ion trap fragmentation is related to the “low mass cut-off”.
- QA10:
Scan range: The lower limit of the applied scan range should ideally be ≤ m/z 50. The lower limit of the applied scan range must not exceed m/z 100 to avoid the production of truncated spetra (instrumental limitations may preclude this, e.g., for instruments relying on ion trapping) wherever possible to include also small mass fragments. The acquired mass range should be given to avoid poor spectral matches due to the presence/absence of low m/z fragments in fragmentation spectra acquired with different scan ranges (see Fig. 7).
- QA11:
Collision energies: Compound-specific breakdown curves should be covered by spectra acquired at multiple collision energies. A spectral series should contain at least 3-5 fragment ion spectra acquired at sufficiently different collision energies within the defined range. With this strategy, libraries are produced that are robust against inter-instrumental collision energy variability (see Figs. 5 and 6). If ramped collision energies are used, these should be clearly labelled as such.
- QA12:
Signal-to-noise: Sample concentration should be sufficiently high to produce fragment ion mass spectra with signal-to-noise ratios >100, to enable reliable acquisition of low-abundant fragment ions.
- QA13:
Saturation: Detector saturation must be avoided for fragment ions, and is only acceptable for precursor ions if the resulting artifacts are removed during curation.
- QA14:
Centroid spectra: Fragment ion mass spectra should be acquired in centroid mode or centroided during export and curation.
- QA15:
Curation: Spectra should be curated, which includes multiple steps of filtering, noise removal, and recalibration, to provide the best quality reference spectra.
- QA16:
Expert review: Spectral series should be reviewed by an expert to identify issues like artifacts, improper noise removal, or truncated spectra, which cannot always be captured automatically.
The curation of acquired tandem mass spectral data is of utmost importance to obtain a high-quality library. Curation efforts may include noise and artifact removal, recalibration of spectra and peak annotations, manual inspection of mass spectra by experienced mass spectrometrists, as well as inter-library comparisons (e.g., [15, 25, 27, 61]). Removal of noise during data processing may lead to losses of spectral information of compounds. Accordingly, processed spectra should be reviewed by experienced mass spectrometrists to check the integrity of data with a special focus on the occurrence of artifacts and processing errors.
Conclusions
Acquisition of tandem mass spectral data requires the physical availability of reference substances and sufficient experimental capacities for acquiring fragment ion mass spectra. Even by joining forces, for instance within HBM4EU and NORMAN initiatives, the acquisition of reference data for ten thousands of compounds is a multi-annual project requiring significant resources. With this document, an outline for harmonized acquisition of suitable spectra for the expansion of public resources is proposed, which balances the consideration of individual instruments and methods at individual laboratories and the comparability of the resulting data. As a result, it is hoped that the next few years will see an increase in the number of environmentally relevant spectra in (open) mass spectral libraries. The question of how to prioritize the compounds for acquisition is being addressed in other activities currently in progress in the HBM4EU project and NORMAN Network and will be communicated separately.
Glossary
Annotation/identification
Capability to assign a signal detected by non-targeted or suspect screening (i.e., a spectrometric descriptor) to a chemical with a given confidence level, by means of a reference library and/or structural elucidation work. Annotation is the act of linking a detected mass spectrometric feature with a chemical. Identification is the act of proving to be the same.
Non-targeted LC–MS
Analytical process for gathering comprehensive information on the composition of a sample. Workflows involve different steps of sample collection, sample preparation, data acquisition and data mining. The fraction of compounds accessible by a certain workflow depends on the characteristics of the individual steps applied. Data-dependent or data-independent acquisition techniques are employed for data acquisition. Detected features are characterized by retention time, MS, and, where possible, MS/MS information to enable annotation.
Targeted LC–MS
Analytical process for gathering specific information on the composition of a sample. Workflows involve different steps of sample collection, sample preparation, data acquisition and data mining. The steps were optimized for a preselected number of molecules. Often selected reaction monitoring techniques are employed for data acquisition. Furthermore, target screening usually involves a reference standard measured in-house under the same analytical conditions such that retention time, MS, and, where possible, MS/MS information is available for identification and confirmation.
Tandem mass spectrometry
Tandem mass spectrometry, also known as MS/MS or MS2, involves multiple steps of mass spectrometry selection, with some form of fragmentation occurring in between the stages. Multiple stages of mass analysis separation can be accomplished with individual mass spectrometer elements separated in space or using a single mass spectrometer with the MS steps separated in time.
Target
A compound that is expected to be included in a sample and of which full mass spectrometric reference data, including MS/MS fragmentation, is available to enable annotation. The reference data is usually acquired with certified reference standards, and is stored in tandem mass spectral databases. The mass spectrometric data is often accompanied by metadata.
Suspect
A compound that is expected to be included in a sample. Typically, the available mass spectrometric data is incomplete and does not allow unequivocal annotation. Often, information on MS/MS fragmentation and retention time is missing or has only been predicted with computational tools.
Known
A detected signal that was annotated to a suspect or target at a certain confidence level.
Unknown
An unannotated signal.
Identification level
An approach for communicating identification confidence. A commonly used classification system in environmental research [2] includes five levels: exact mass—unequivocal molecular formula—tentative candidate—probable structure—confirmed structure.
Tandem mass spectral database
An organized collection of tandem mass spectral data which comes bundled with a management system. The database management system is a software application that interacts with the user, other applications, and the database itself to capture and analyze data. Tandem mass spectra are typically acquired from certified reference compounds. Spectral information is processed prior to storage in a library.
Tandem mass spectral library
A curated and annotated collection of mass spectra acquired from certified reference compounds. Curation efforts may include manual inspection of mass spectra by experienced mass spectrometrists, noise and artifact removal, recalibration of spectra and peak annotations, as well as inter-library comparisons. The mass spectrometric data is often accompanied by metadata.
Availability of data and materials
Datasets generated and analyzed during the current study will be available in MassBank (https://massbank.eu/MassBank/) in a HBM4EU_INTERLAB_2019 folder and for direct download via GitHub (https://github.com/MassBank/MassBank-data/tree/master/HBM4EU_INTERLAB_2019)
The WRTMD is available from John Wiley & Sons, Inc. [53].
Abbreviations
- AGC:
-
Automatic gain control
- amp:
-
Average match probability
- CID:
-
Collision-induced dissociation
- Eawag:
-
Swiss Federal Institute of Aquatic Science and Technology
- ESI:
-
Electrospray ionization
- FTICR:
-
Fourier transform ion cyclotron resonance
- GNPS:
-
Global Natural Product Social Molecular Networking
- HBM4EU:
-
Human Biomonitoring for Europe project
- HCD:
-
Higher-energy collisional dissociation
- HMDB:
-
Human Metabolome Database
- HRMS:
-
High-resolution mass spectrometry
- IT:
-
Ion trap
- LC:
-
Liquid chromatography
- LIT:
-
Linear ion trap
- m/z:
-
Mass-to-charge ratio
- MoNA:
-
MassBank of North America
- mp:
-
Reference spectrum-specific match probability
- MS:
-
Mass spectrometry
- MS/MS, MS2:
-
Tandem mass spectrometry
- NCE:
-
Normalized collision energy
- NORMAN:
-
Network of reference laboratories, research centers and related organizations for monitoring of emerging environmental substances
- Q:
-
Quadrupole
- QA:
-
Quality assurance
- QC:
-
Quality control
- QqTOF:
-
Quadrupole–quadrupole time of flight instrument
- ramp:
-
Relative average match probability
- ReSpect:
-
RIKEN MSn spectral database for phytochemicals
- TEDB:
-
Toxic Exposome Database
- UFZ:
-
Helmholtz Centre for Environmental Research
- WRTMD:
-
Wiley Registry of Tandem Mass Spectra Database
References
Hollender J, Schymanski EL, Singer HP, Ferguson PL (2017) Nontarget screening with high resolution mass spectrometry in the environment: ready to go? Environ Sci Technol 51:11505–11512. https://doi.org/10.1021/acs.est.7b02184
Schymanski EL, Jeon J, Gulde R, Fenner K, Ruff M, Singer HP, Hollender J (2014) Identifying small molecules via high resolution mass spectrometry: communicating confidence. Environ Sci Technol 48:2097–2098. https://doi.org/10.1021/es5002105
Blaženović I, Kind T, Ji J, Fiehn O (2018) Software tools and approaches for compound identification of LC–MS/MS data in metabolomics. Metabolites 8:31. https://doi.org/10.3390/metabo8020031
Schymanski EL, Ruttkies C, Krauss M, Brouard C, Kind T, Dührkop K, Allen F, Vaniya A, Verdegem D, Böcker S, Rousu J, Shen H, Tsugawa H, Sajed T, Fiehn O, Ghesquière B, Neumann S (2017) Critical assessment of small molecule identification 2016: automated methods. J Cheminform 9:22. https://doi.org/10.1186/s13321-017-0207-1
Ruttkies C, Schymanski EL, Wolf S, Hollender J, Neumann S (2016) MetFrag relaunched: incorporating strategies beyond in silico fragmentation. J Cheminform 8:3. https://doi.org/10.1186/s13321-016-0115-9
Blaženović I, Kind T, Torbašinović H, Obrenović S, Mehta SS, Tsugawa H, Wermuth T, Schauer N, Jahn M, Biedendieck R, Jahn D, Fiehn O (2017) Comprehensive comparison of in silico MS/MS fragmentation tools of the CASMI contest: database boosting is needed to achieve 93% accuracy. J Cheminform 9:32. https://doi.org/10.1186/s13321-017-0219-x
Allen F, Pon A, Wilson M, Greiner R, Wishart D (2014) CFM-ID: a web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res 42:W94–W99. https://doi.org/10.1093/nar/gku436
Djoumbou-Feunang Y, Pon A, Karu N, Zheng J, Li C, Arndt D, Gautam M, Allen F, Wishart DS (2019) CFM-ID 3.0: Significantly improved ESI-MS/MS prediction and compound identification. Metabolites 9:72. https://doi.org/10.3390/metabo9040072
Bade R, Bijlsma L, Miller TH, Barron LP, Sancho JV, Hernández F (2015) Suspect screening of large numbers of emerging contaminants in environmental waters using artificial neural networks for chromatographic retention time prediction and high resolution mass spectrometry data analysis. Sci Total Environ 538:934–941. https://doi.org/10.1016/j.scitotenv.2015.08.078
Creek DJ, Jankevics A, Breitling R, Watson DG, Barrett MP, Burgess KEV (2011) Toward global metabolomics analysis with hydrophilic interaction liquid chromatography-mass spectrometry: improved metabolite identification by retention time prediction. Anal Chem 83:8703–8710. https://doi.org/10.1021/ac2021823
Stanstrup J, Neumann S, Vrhovšek U (2015) PredRet: prediction of retention time by direct mapping between multiple chromatographic systems. Anal Chem 87:9421–9428. https://doi.org/10.1021/acs.analchem.5b02287
Goryński K, Bojko B, Nowaczyk A, Buciński A, Pawliszyn J, Kaliszan R (2013) Quantitative structure–retention relationships models for prediction of high performance liquid chromatography retention time of small molecules: endogenous metabolites and banned compounds. Anal Chim Acta 797:13–19. https://doi.org/10.1016/j.aca.2013.08.025
Schymanski EL, Williams AJ (2017) Open science for identifying “known unknown” chemicals. Environ Sci Technol 51:5357–5359. https://doi.org/10.1021/acs.est.7b01908
Sumner LW, Amberg A, Barrett D, Beale MH, Beger R, Daykin CA, Fan TW-M, Fiehn O, Goodacre R, Griffin JL, Hankemeier T, Hardy N, Harnly J, Higashi R, Kopka J, Lane AN, Lindon JC, Marriott P, Nicholls AW, Reily MD, Thaden JJ, Viant MR (2007) Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 3:211–221. https://doi.org/10.1007/s11306-007-0082-2
Stravs MA, Schymanski EL, Singer HP, Hollender J (2013) Automatic recalibration and processing of tandem mass spectra using formula annotation: recalibration and processing of MS/MS spectra. J Mass Spectrom 48:89–99. https://doi.org/10.1002/jms.3131
Vinaixa M, Schymanski EL, Neumann S, Navarro M, Salek RM, Yanes O (2016) Mass spectral databases for LC/MS- and GC/MS-based metabolomics: state of the field and future prospects. TrAC Trends Anal Chem 78:23–35. https://doi.org/10.1016/j.trac.2015.09.005
Frainay C, Schymanski E, Neumann S, Merlet B, Salek R, Jourdan F, Yanes O (2018) Mind the gap: mapping mass spectral databases in genome-scale metabolic networks reveals poorly covered areas. Metabolites 8:51. https://doi.org/10.3390/metabo8030051
Oberacher H, Arnhard K (2016) Current status of non-targeted liquid chromatography-tandem mass spectrometry in forensic toxicology. TrAC Trends Anal Chem 84:94–105. https://doi.org/10.1016/j.trac.2015.12.019
Oberacher H, Arnhard K (2015) Compound identification in forensic toxicological analysis with untargeted LC–MS-based techniques. Bioanalysis 7:2825–2840. https://doi.org/10.4155/bio.15.193
Kind T, Tsugawa H, Cajka T, Ma Y, Lai Z, Mehta SS, Wohlgemuth G, Barupal DK, Showalter MR, Arita M, Fiehn O (2018) Identification of small molecules using accurate mass MS/MS search. Mass Spectrom Rev 37:513–532. https://doi.org/10.1002/mas.21535
Milman BL, Zhurkovich IK (2016) Mass spectral libraries: a statistical review of the visible use. TrAC Trends Anal Chem 80:636–640. https://doi.org/10.1016/j.trac.2016.04.024
Cooper BT, Yan X, Simón-Manso Y, Tchekhovskoi DV, Mirokhin YA, Stein SE (2019) Hybrid search: a method for identifying metabolites absent from tandem mass spectrometry libraries. Anal Chem 91(21):13924–13932. https://doi.org/10.1021/acs.analchem.9b03415
Stein S (2012) Mass spectral reference libraries: an ever-expanding resource for chemical identification. Anal Chem 84:7274–7282. https://doi.org/10.1021/ac301205z
Horai H, Arita M, Kanaya S, Nihei Y, Ikeda T, Suwa K, Ojima Y, Tanaka K, Tanaka S, Aoshima K, Oda Y, Kakazu Y, Kusano M, Tohge T, Matsuda F, Sawada Y, Hirai MY, Nakanishi H, Ikeda K, Akimoto N, Maoka T, Takahashi H, Ara T, Sakurai N, Suzuki H, Shibata D, Neumann S, Iida T, Tanaka K, Funatsu K, Matsuura F, Soga T, Taguchi R, Saito K, Nishioka T (2010) MassBank: a public repository for sharing mass spectral data for life sciences. J Mass Spectrom 45:703–714. https://doi.org/10.1002/jms.1777
Wallace WE, Ji W, Tchekhovskoi DV, Phinney KW, Stein SE (2017) Mass spectral library quality assurance by inter-library comparison. J Am Soc Mass Spectrom 28:733–738. https://doi.org/10.1007/s13361-016-1589-4
Yang X, Neta P, Stein SE (2014) Quality control for building libraries from electrospray ionization tandem mass spectra. Anal Chem 86:6393–6400. https://doi.org/10.1021/ac500711m
Oberacher H, Reinstadler V, Kreidl M, Stravs M, Hollender J, Schymanski E (2018) Annotating nontargeted LC-HRMS/MS data with two complementary tandem mass spectral libraries. Metabolites 9:3. https://doi.org/10.3390/metabo9010003
Wishart DS, Feunang YD, Marcu A, Guo AC, Liang K, Vázquez-Fresno R, Sajed T, Johnson D, Li C, Karu N, Sayeeda Z, Lo E, Assempour N, Berjanskii M, Singhal S, Arndt D, Liang Y, Badran H, Grant J, Serra-Cayuela A, Liu Y, Mandal R, Neveu V, Pon A, Knox C, Wilson M, Manach C, Scalbert A (2018) HMDB 4.0: the Human Metabolome Database for 2018. Nucleic Acids Res 46:D608–D617. https://doi.org/10.1093/nar/gkx1089
FiehnLab (2019) MassBank of North America. http://mona.fiehnlab.ucdavis.edu/. Accessed 14 Mar 2019
Wishart DS, Jewison T, Guo AC, Wilson M, Knox C, Liu Y, Djoumbou Y, Mandal R, Aziat F, Dong E, Bouatra S, Sinelnikov I, Arndt D, Xia J, Liu P, Yallou F, Bjorndahl T, Perez-Pineiro R, Eisner R, Allen F, Neveu V, Greiner R, Scalbert A (2013) HMDB 3.0—the Human Metabolome Database in 2013. Nucleic Acids Res 41:D801–D807. https://doi.org/10.1093/nar/gks1065
Wang M, Carver JJ, Phelan VV, Sanchez LM, Garg N, Peng Y, Nguyen DD, Watrous J, Kapono CA, Luzzatto-Knaan T, Porto C, Bouslimani A, Melnik AV, Meehan MJ, Liu W-T, Crüsemann M, Boudreau PD, Esquenazi E, Sandoval-Calderón M, Kersten RD, Pace LA, Quinn RA, Duncan KR, Hsu C-C, Floros DJ, Gavilan RG, Kleigrewe K, Northen T, Dutton RJ, Parrot D, Carlson EE, Aigle B, Michelsen CF, Jelsbak L, Sohlenkamp C, Pevzner P, Edlund A, McLean J, Piel J, Murphy BT, Gerwick L, Liaw C-C, Yang Y-L, Humpf H-U, Maansson M, Keyzers RA, Sims AC, Johnson AR, Sidebottom AM, Sedio BE, Klitgaard A, Larson CB, P CAB, Torres-Mendoza D, Gonzalez DJ, Silva DB, Marques LM, Demarque DP, Pociute E, O’Neill EC, Briand E, Helfrich EJN, Granatosky EA, Glukhov E, Ryffel F, Houson H, Mohimani H, Kharbush JJ, Zeng Y, Vorholt JA, Kurita KL, Charusanti P, McPhail KL, Nielsen KF, Vuong L, Elfeki M, Traxler MF, Engene N, Koyama N, Vining OB, Baric R, Silva RR, Mascuch SJ, Tomasi S, Jenkins S, Macherla V, Hoffman T, Agarwal V, Williams PG, Dai J, Neupane R, Gurr J, Rodríguez AMC, Lamsa A, Zhang C, Dorrestein K, Duggan BM, Almaliti J, Allard P-M, Phapale P, Nothias L-F, Alexandrov T, Litaudon M, Wolfender J-L, Kyle JE, Metz TO, Peryea T, Nguyen D-T, VanLeer D, Shinn P, Jadhav A, Müller R, Waters KM, Shi W, Liu X, Zhang L, Knight R, Jensen PR, Palsson BO, Pogliano K, Linington RG, Gutiérrez M, Lopes NP, Gerwick WH, Moore BS, Dorrestein PC, Bandeira N (2016) Sharing and community curation of mass spectrometry data with global natural products social molecular networking. Nat Biotechnol 34:828–837. https://doi.org/10.1038/nbt.3597
Sawada Y, Nakabayashi R, Yamada Y, Suzuki M, Sato M, Sakata A, Akiyama K, Sakurai T, Matsuda F, Aoki T, Hirai MY, Saito K (2012) RIKEN tandem mass spectral database (ReSpect) for phytochemicals: a plant-specific MS/MS-based data resource and database. Phytochemistry 82:38–45. https://doi.org/10.1016/j.phytochem.2012.07.007
Lam H (2011) Building and Searching tandem mass spectral libraries for peptide identification. Mol Cell Proteomics 10(R111):008565. https://doi.org/10.1074/mcp.R111.008565
Oberacher H, Pavlic M, Libiseller K, Schubert B, Sulyok M, Schuhmacher R, Csaszar E, Köfeler HC (2009) On the inter-instrument and the inter-laboratory transferability of a tandem mass spectral reference library: 2. Optimization and characterization of the search algorithm: about an advanced search algorithm for tandem mass spectral reference libraries. J Mass Spectrom 44:494–502. https://doi.org/10.1002/jms.1525
Pavlic M, Libiseller K, Oberacher H (2006) Combined use of ESI–QqTOF-MS and ESI–QqTOF-MS/MS with mass-spectral library search for qualitative analysis of drugs. Anal Bioanal Chem 386:69–82. https://doi.org/10.1007/s00216-006-0634-8
Oberacher H, Whitley G, Berger B, Weinmann W (2013) Testing an alternative search algorithm for compound identification with the ‘Wiley Registry of Tandem Mass Spectral Data, MSforID’: an alternative search algorithm for the Wiley Registry MSMS. J Mass Spectrom 48:497–504. https://doi.org/10.1002/jms.3185
Mylonas R, Mauron Y, Masselot A, Binz P-A, Budin N, Fathi M, Viette V, Hochstrasser DF, Lisacek F (2009) X-Rank: a robust algorithm for small molecule identification using tandem mass spectrometry. Anal Chem 81:7604–7610. https://doi.org/10.1021/ac900954d
Nesvizhskii AI, Vitek O, Aebersold R (2007) Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat Methods 4:787–797. https://doi.org/10.1038/nmeth1088
Scheubert K, Hufsky F, Petras D, Wang M, Nothias L-F, Dührkop K, Bandeira N, Dorrestein PC, Böcker S (2017) Significance estimation for large scale metabolomics annotations by spectral matching. Nat Commun 8:1494. https://doi.org/10.1038/s41467-017-01318-5
Ichou F, Schwarzenberg A, Lesage D, Alves S, Junot C, Machuron-Mandard X, Tabet J-C (2014) Comparison of the activation time effects and the internal energy distributions for the CID, PQD and HCD excitation modes: theoretical comparison of CID, PQD and HCD. J Mass Spectrom 49:498–508. https://doi.org/10.1002/jms.3365
NORMAN Network NORMAN Suspect List Exchange. https://www.norman-network.com/nds/SLE/. Accessed 9 Jun 2019
Williams AJ, Grulke CM, Edwards J, McEachran AD, Mansouri K, Baker NC, Patlewicz G, Shah I, Wambaugh JF, Judson RS, Richard AM (2017) The CompTox chemistry dashboard: a community data resource for environmental chemistry. J Cheminform 9:61. https://doi.org/10.1186/s13321-017-0247-6
NORMAN Network (2019) NORMAN suspect list exchange database SusDat. https://www.norman-network.com/nds/susdat/. Accessed 15 Mar 2019
Wishart DS, Feunang YD, Guo AC, Lo EJ, Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z, Assempour N, Iynkkaran I, Liu Y, Maciejewski A, Gale N, Wilson A, Chin L, Cummings R, Le D, Pon A, Knox C, Wilson M (2018) DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 46:D1074–D1082. https://doi.org/10.1093/nar/gkx1037
Wishart D, Arndt D, Pon A, Sajed T, Guo AC, Djoumbou Y, Knox C, Wilson M, Liang Y, Grant J, Liu Y, Goldansaz SA, Rappaport SM (2015) T3DB: the toxic exposome database. Nucleic Acids Res 43:D928–D934. https://doi.org/10.1093/nar/gku1004
Neveu V, Moussy A, Rouaix H, Wedekind R, Pon A, Knox C, Wishart DS, Scalbert A (2017) Exposome-Explorer: a manually-curated database on biomarkers of exposure to dietary and environmental factors. Nucleic Acids Res 45:D979–D984. https://doi.org/10.1093/nar/gkw980
HighChem LLC (2019) mzCloud advanced mass spectral database. https://www.mzcloud.org/. Accessed 14 Mar 2019
NORMAN Network, MassBank Consortium (2019) MassBank EU: European MassBank (NORMAN MassBank). https://massbank.eu/MassBank/. Accessed 15 Mar 2019
Oberacher HM (2019) WRTMD or MSforID: Tandem mass spectral identification of small molecules. https://msforid.com/. Accessed 20 Dec 2019
Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, Li Q, Shoemaker BA, Thiessen PA, Yu B, Zaslavsky L, Zhang J, Bolton EE (2019) PubChem 2019 update: improved access to chemical data. Nucleic Acids Res 47:D1102–D1109. https://doi.org/10.1093/nar/gky1033
Daylight Chemical Information Systems, Inc. (2008) SMILES—a simplified chemical language. http://www.daylight.com/dayhtml/doc/theory/theory.smiles.html. Accessed 13 Apr 2019
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR (2011) Open Babel: an open chemical toolbox. J Cheminform 3:33. https://doi.org/10.1186/1758-2946-3-33
Oberacher HM (2011) The Wiley registry of tandem mass spectral data, MSforID., 1st edn. John Wiley & Sons, Hoboken
Peisl BYL, Schymanski EL, Wilmes P (2018) Dark matter in host-microbiome metabolomics: tackling the unknowns—a review. Anal Chim Acta 1037:13–27. https://doi.org/10.1016/j.aca.2017.12.034
Gaston L, Lapworth DJ, Stuart M, Arnscheidt J (2019) Prioritization approaches for substances of emerging concern in groundwater: a critical review. Environ Sci Technol 53:6107–6122. https://doi.org/10.1021/acs.est.8b04490
Götz CW, Stamm C, Fenner K, Singer H, Schärer M, Hollender J (2010) Targeting aquatic microcontaminants for monitoring: exposure categorization and application to the Swiss situation. Environ Sci Pollut Res 17:341–354. https://doi.org/10.1007/s11356-009-0167-8
Little JL, Cleven CD, Brown SD (2011) Identification of “known unknowns” utilizing accurate mass data and chemical abstracts service databases. J Am Soc Mass Spectrom 22:348–359. https://doi.org/10.1007/s13361-010-0034-3
Oberacher H, Pitterl F, Siapi E, Steele BR, Letzel T, Grosse S, Poschner B, Tagliaro F, Gottardo R, Chacko SA, Josephs JL (2012) On the inter-instrument and the inter-laboratory transferability of a tandem mass spectral reference library. 3. Focus on ion trap and upfront CID: on the transferability of a tandem mass spectral reference library. J Mass Spectrom 47:263–270. https://doi.org/10.1002/jms.2961
Boyd RK, Basic C, Bethem RA (2013) Trace quantitative analysis by mass spectrometry. Wiley, Hoboken
McEachran AD, Mansouri K, Grulke C, Schymanski EL, Ruttkies C, Williams AJ (2018) “MS-Ready” structures for non-targeted high-resolution mass spectrometry screening studies. J Cheminform 10:45. https://doi.org/10.1186/s13321-018-0299-2
Damont A, Olivier M-F, Warnet A, Lyan B, Pujos-Guillot E, Jamin EL, Debrauwer L, Bernillon S, Junot C, Tabet J-C, Fenaille F (2019) Proposal for a chemically consistent way to annotate ions arising from the analysis of reference compounds under ESI conditions: a prerequisite to proper mass spectral database constitution in metabolomics. J Mass Spectrom 54:567–582. https://doi.org/10.1002/jms.4372
Acknowledgements
LD thanks Alyssa Bouville for her assisting in acquiring tandem mass spectral data.
Funding
The authors acknowledge financial support by the HBM4EU project. HBM4EU has received funding from the European Union’s Horizon 2020 research and innovation programme under Grant Agreement No. 733032. This text has been modified and extended for publication from Deliverable AD16.4 as part of WP16 within the HBM4EU project. ELS acknowledges funding from the Luxembourg National Research Fund (FNR) for project A18/BM/12341006. MS acknowledges financial support by the University of Innsbruck.
Author information
Authors and Affiliations
Contributions
HO, JPA, LD, MK, AC, FF, ML, ELS elaborated conception and design of this study. HO, MS, JPA, YG, LD, ELJ, TS, MK, AC, NCC, KR, AD, FF, ELS were involved in data acquisition and analysis. HO and ELS wrote a first version of the manuscript. All authors were involved in drafting and revising the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Extended description of experimental settings.
Additional file 2.
Reference compounds for producing tandem mass spectral libraries.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Oberacher, H., Sasse, M., Antignac, JP. et al. A European proposal for quality control and quality assurance of tandem mass spectral libraries. Environ Sci Eur 32, 43 (2020). https://doi.org/10.1186/s12302-020-00314-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12302-020-00314-9